Що визначає, як швидкість реакції реагує на термодинамічні рушійні сили? І чому деякі реакції різко прискорюються навіть за невеликих змін цих сил, тоді як інші залишаються майже нечутливими? На ці запитання спробували відповісти теоретичні хіміки з Німеччини, запропонувавши нову концептуальну основу для моделювання реакційних ландшафтів хімічних процесів.
Розуміння ролі зовнішніх факторів у хімічних реакціях є ключовим як для теоретичних, так і для експериментальних досліджень. Глибше усвідомлення цих факторів дозволяє хімікам спрямовувати реакції у бік бажаних продуктів, підвищувати вихід і зменшувати кількість побічних процесів.
Пошук стабільності: термодинамічний підхід
Хімічні реакції можна розглядати як безперервний пошук стабільності. З термодинамічної точки зору, стабільність вимірюється вільною енергією Гіббса, і реакція зазвичай відбувається у напрямку її зниження. Проте цей шлях не є єдиним — система може проходити через кілька «субоптимальних» проміжних станів, залежно від зовнішніх умов.
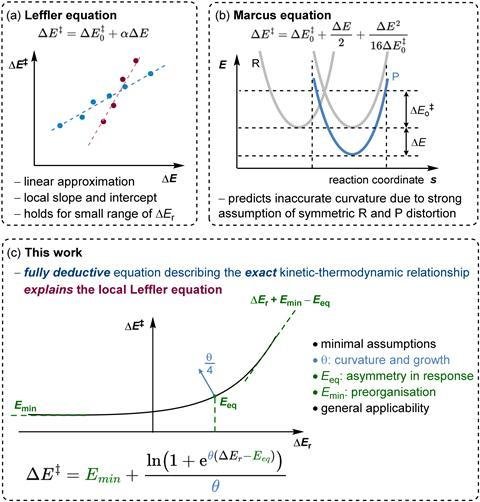
Класичні моделі, такі як рівняння Леффлера чи рівняння Маркуса, намагалися описати, як швидкість реакції залежить від зовнішніх рушійних сил, щоб визначити найвигідніший шлях реакції. Проте ці моделі мають обмеження у колі термодинамічних ситуацій, які вони можуть адекватно описати. Більш сучасні статистичні підходи охоплюють ширший діапазон реакцій, однак потребують великої кількості специфічних параметрів, що ускладнює їх тлумачення.
Нова формула з одним параметром
Тепер Едуардо Гарсія-Паділья та Гуаньці Цю з Інституту досліджень вугілля Товариства Макса Планка (Німеччина) запропонували модель, що пов’язує енергію активації реакції (ΔE‡) з її загальною енергією реакції (ΔEr). Їхнє рівняння використовує лише один параметр — фактор кривизни θ, який відображає стабільність перехідного стану та його залежність від конкурентних чинників, що впливають на реакцію.
На відміну від попередніх підходів, ця модель описує основні властивості хімічних реакцій без потреби у жорстких припущеннях. Таким чином, вона зберігає точність статистичних методів, але при цьому залишається такою ж простою, як класичні рівняння.
Гарсія-Паділья прогнозує, що їхня модель допоможе хімікам відкривати нові реакційні шляхи, виявляючи приховані зв’язки між силами, які керують хімічними перетвореннями.
«Деякі параметри, наприклад, показують, що існує мінімальна енергія активації, яку неможливо подолати лише тим, що реакція стає більш вигідною термодинамічно. Це вже дає хімікам більше контролю над процесом, ніж припущення про лінійну залежність між вільною енергією та швидкістю реакції», — пояснює він.
У майбутньому, за словами Гарсії-Падільї, передбачення параметрів моделі для певного сімейства реакцій дозволить хімікам оцінювати, наскільки сильно реакція реагуватиме на зовнішній вплив, і регулювати цей параметр для підвищення селективності між можливими напрямками процесу.
Перспективи поєднання з машинним навчанням
Дослідникм також вважають, що машинне навчання стане інструментом для прогнозування параметрів моделі у межах певних груп реакцій:
«Такі комбінації, коли машинне навчання не передбачає саму швидкість реакції, а оцінює фізично значущі параметри, виглядають дуже перспективними. Навіть якщо нейромережа працює як “чорна скринька”, перевага полягає в тому, що отримані параметри легко перевірити на коректність».
Погляд з боку експертів
На думку Марко Мартінеса з Університету Макмастера (Онтаріо, Канада), фахівця з теоретичної хімії та моделювання реакційної здатності,
«Дослідження є своєчасним і вагомим внеском у галузі обчислювальної хімії та дизайну реакцій. Визначення енергетичних бар’єрів зазвичай потребує оптимізації станів реагентів і перехідних структур, що є трудомістким і сильно залежить від досвіду дослідника. Тому такий підхід непридатний для високопродуктивного скринінгу. Натомість запропонована модель може бути параметризована на основі вже наявних даних, що робить її придатною для автоматизованих розрахунків, поєднуючи низькі обчислювальні витрати з фізично осмисленими прогнозами».
Водночас, додає Мартінес, модель потребує додаткової перевірки:
«Її параметри пов’язані з хімічно інтерпретованими поняттями, але не мають однозначного зв’язку з властивостями реагентів і продуктів. Крім того, її ефективність варто перевірити для реакцій із високою асинхронністю або для систем із кількома конкуруючими шляхами, що мають схожі бар’єри».